
Standard Operating Procedure (SOP) for Handling of Data Integrity Observations (DIO) and Data Integrity Breach (DIB). Data Integrity is the extent to which all data are complete, consistent, truthful, traceable and accurate through the data life-cycle.
Procedure For Handling of Data Integrity Observations
1.0 PURPOSE
-
- The purpose of this Standard Operating Procedure (SOP) is;
-
- To outlines, the process for Handling of Data Integrity Observations (DIO) in order to ensure that each such observation is identified, documented, investigated and concluded appropriately along with with implementation of any corrective and/or preventive actions, which will eliminate/reduce chances of recurrence of the same issue.
2.0 SCOPE
-
- This SOP shall be applicable as such to handle the Data Integrity Observation (DIO) discovered concerning any cGxP process carried out at the pharmaceutical manufacturing plant.
3.0 RESPONSIBILITY
-
-
Data / Record generator shall be responsible for :
-
-
- Cooperation during any Data Integrity Observation investigation.
-
- Reporting all original data as per the respective SOP(s).
-
- Submission of the supporting data to the data reviewer.
-
- Correcting any deficiency observations identified by the reviewer.
-
- Timely reporting all quality impacting events such as deviations/incident, OOS, OOT, etc. as per the respective SOP(s).
-
-
Data / Document reviewer shall be responsible for :
-
-
- Informing Data/Record Generator of any deficiencies identified during the review.
-
- Ensuring adequacy of compliance for the identified observations.
-
- Ensuring all quality events such as incident/ deviation, OOS, OOT, etc. Investigated and documented in the reviewing Datasheet/worksheets and in accordance with the current version of applicable SOP requirements.
-
- Good documentation practices are followed and in compliance with the procedures.
-
-
Head of department /Section Head or designee shall be responsible for :
-
-
- Data storage and traceability.
Related: SOP for Document Management System
-
- Identification of resource constraints that could lead to a breach in data reliability/integrity and communicate to staff that resource constrains should never lead to a breach in data integrity.
-
- Ensuring personnel responsible for data generation/recording and data review adequately trained in procedures and policies relating to assurance of DI.
-
- Full support and assistance during any DIO investigation.
-
-
Head QA shall be responsible for :
-
-
- Ensuring personnel responsible for data generation/recording, data review, data archival, equipment and systems, supervision, management and oversight related to cGxP processes are adequately trained in procedures and policies relating to assurance of Data Integrity.
-
- Identification of resource constraints that could lead to a breach in data reliability/integrity and communicate to staff that resource constrains should never lead to a breach in data integrity.
-
- Focus on prevention actions that support data reliability.
-
- Development of the processes for employees to report any breaches in data reliability to senior management.
-
- Ensuring adequate information flow between staff at all levels.
-
- Encouragement to take an open door approach and execution of the appropriate actions as per open door privileges for employees.
-
- Periodic risk assessment of the Quality System regarding potential data integrity issues.
-
- Leadership during any Data Integrity Observation investigation.
-
- To build systems and controls which prevent employees from altering or falsifying the data.
-
-
Head of Personnel and Administration (PA) department shall be responsible for :
-
-
- To participate/support Data Integrity Observation investigation.
-
- To take disciplinary action in case Data Integrity Observation turns into DIB.
-
-
Plant Management and Corporate Quality shall be responsible for :
-
-
- For making staff aware of the relevance of data reliability and its importance.
-
- To create a work environment in which staff is encouraged to communicate failures and mistakes related to data reliability so that corrective action can be taken.
-
- To build systems and controls which prevent employees from altering or falsifying the data.
4.0 PROCEDURE FOR HANDLING OF DATA INTEGRITY OBSERVATIONS
-
-
Handling of Data Integrity Observations (DIO):
-
-
- Data Integrity Observations (DIOs): A DIO may be identified through one or more of the following mechanisms, but not limited to:
-
-
- Regulatory Agency Investigation
-
-
-
- Regulatory Compliance Review
-
-
-
- Quality Assurance Review
-
-
-
- Others
-
Flow Chart – Handling of Data Integrity Observation (DIO)
")
")
")
-
- For every Data Integrity Observation (DIO) event, a Quality Alert shall send by the QA Head / Head CQ (wherever applicable).
-
- Consider the DIO events as an “Incident” for processing in the Quality Management Systems.
-
- Hence, for each DIO event, Log the incident as per the current version of the respective SOP
-
- A site leadership team consisting of minimum QA Head, respective department head, PA Head and Plant Head to investigate the DIO.
-
- Include the corporate quality representative included if needed.
-
-
Major Steps in the DIO Investigation Process:
-
-
- An impact/risk assessment of the DIO shall be performed to determine any potential for adverse impact on product quality, efficacy, patient safety and initiate immediate corrections/control measures over impacted product(s), particularly any products released into the market (e.g. quarantine, temporary suspension of relevant product(s)/manufacturing operations, etc.).
-
- Investigation & Risk Assessment of DIO: Investigation of DIO shall consist of the following steps:
-
- Step 1: Ascertaining whether the “DIO” is a Data Integrity Breach (DIB) or not, based on the following (but not limited to):
-
-
- Interviews of the personnel involved and immediate supervisors/subordinates.
-
-
-
- Document the records of the interviews conducted as part of the investigations.
-
-
-
- Review of past behavioral attributes of the personnel involved.
-
-
-
- Review of past records of activities performed by the personnel involved (e.g. manufacturing, testing, document preparation, etc.).
-
-
-
- Work allocations review of personnel involved.
-
-
-
- Review of other equipment/instruments/systems operated used by the personnel involved in the said DIO.
-
-
-
- Evaluate the involvement of other related personnel (e.g. superiors/subordinates) etc.
-
-
- If the DIO is concluded as “not a Data Integrity Breach (DIB)” (i.e. only as a non-compliance of Good Documentation Practices (GDP) or Good Manufacturing Practices (GMP) and does not impact data integrity), then further steps of the Incident (e.g. Root Cause Analysis, Risk Assessment, CAPA/Other Actions generation and processing, etc.) shall be processed, as per applicable SOP of Incident (i.e., similar to all other Incident) to remediate the event.
-
-
If the DIO is concluded as a “DIB”, Follow the following steps :
-
-
- Step 2: The appointed DIO Lead Investigator shall carry out a detailed Investigational Analysis, including Root-Cause Analysis and Risk Assessment on two aspects and these activities shall be conducted in parallel:
-
- Product Quality, Efficacy, Patient Safety Aspects
-
- Personnel Perspective
-
- Incident – Investigation – Product Quality, Efficacy, and Patient Safety Aspects:
-
- Based on the nature of the DIO, the DIO Investigator (with the concern of Head QA) may extend the investigation to :
-
- Other batches tested/ manufactured by the personnel involved in the said DIO.
-
- Documents/records prepared by the personnel involved in the said DIO.
-
- Other equipment/instruments/ systems operated/used by the personnel involved in the said DIO.
-
- Regulatory filings/approved Marketing Authorization.
-
- Using the investigation outcomes, the level of risk on product quality/patient safety shall be calculated as either Critical / Major / Minor and this assessment shall be made based on the “Severity” and “Probability of Occurrence” of the DIO event and corresponding “State of Controls/mitigation plan”.
Related: SOP for Quality Risk Management
-
- The investigational outcomes and risk classification shall be shared by the QA Head with Regulatory Affairs (wherever applicable), who shall ascertain the need to inform the relevant Regulatory Agency about the DIO event in accordance with prevailing procedures.
-
- Additionally, the PA head (with the concern of Plant Head) shall decide on the actions against the personnel involved in the DIB, based on the risk category and company’s procedures.
-
-
Trend Analysis of Data Integrity Observations:
-
-
- Perform the Trend analysis of DIBs at (at least annually) by QA, either as a part of the Quality Review Meeting or as a separate activity.
-
- Carryout the Trend analysis on one or more of the following parameters (but not limited to): Nature of DIB, DIB Risk Classification and Root Cause Category.
-
- Identify any adverse trends, the effectiveness of CAPA implemented, assessed and additional action (if needed) proposed to address observed repetitive patterns of DIBs.
5.0 REFERENCES FOR DATA INTEGRITY SOP
-
- PIC/S Guideline: Good Practices for Data Integrity in Regulated GMP / GDP Environments. (Aug 2016)
-
- MHRA Guideline: MHRA GMP Data Integrity Definitions and Guidance for Industry – March 2015
6.0 ABBREVIATIONS AND DEFINITIONS
-
- CAPA : Corrective and Preventive Action
-
- DIO : Data Integrity Observation
-
- DIO : Data Integrity Breach
-
- GMP : Good Manufacturing Practices
-
- GDP : Good Documentation Practices
-
- OOS : Out of Specification
-
- OOT : Out of Trend
| Term | Definition |
| Data Integrity | The extent to which all data are complete, consistent, truthful, traceable and accurate through the data life-cycle. |
| Data/Document Reviewer | The primary reviewer responsible for the checking of the accuracy, reliability, validity and completeness of data/records generated/reported during cGxP processes. |
| Data/Record Generator | Anyone involved in the generation/recording of cGxP data. |
| DIB | Data Integrity Breach: Any observation which is confirmed to be compromising with the said definition of Data Integrity is called a Data Integrity Breach. |
| DIO | Data Integrity Observation: Any observation which is suspected to compromise with the said definition of Data Integrity is called a Data Integrity Observation. |
| Dynamic Record | Records in a dynamic formats, such as electronic records, that allows for an interactive relationship between the user and the record content. |
| Electronic Record | Electronic Data used in lieu of paper cGxP records. |
Fraud |
Deliberate falsification. |
| Improper Practice | Unjustified omission, manipulation, or alteration of procedures or data that bypasses the required parameters, making results appear acceptable |
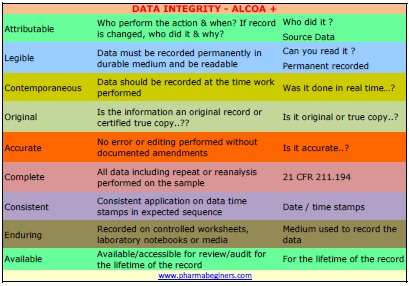
| Attributable | Attributable means information is captured in the record so that it is uniquely identified as executed by the originator of the data (e.g. a person, computer system). |
| Legible | The terms legible and traceable and permanent refer to the requirements that data are readable, understandable and allow a clear picture of the sequencing of steps or events in the record. |
| Contemporaneous | Contemporaneous is process of documentation (on paper or electronically) at the time of an activity. |
| Original | Original data includes the first or source capture of data or information and all subsequent data required to fully reconstruct the conduct of the cGxP activity. |
Accurate |
The term “accurate” means data are correct, truthful, valid and reliable. |
| Complete | All data from an analysis, including any data generated before a problem is observed, data generated after repeat part or all of the work or reanalysis performed on the sample. For hybrid systems, the paper output must be linked to the underlying electronic records used to produce it. |
| Consistent | All elements of the analysis, such as the sequence of events, follow on and data files are date (all processes) and time (when using a hybrid or electronic systems) stamped in the expected order |
| Enduring | Recorded on authorized media e.g. laboratory notebooks, numbered worksheets, for which there is accountability or electronic media. Not recorded on scrap paper or any other media which later discarded e.g. the backs of envelopes, laboratory coat sleeves or Post-It notes |
| Available | The complete collection of records can be accessed or retrieved for review and audit or inspection over the lifetime of the record. |
Pingback: Data Integrity: A Crucial Aspect of Pharmaceutical Quality -
Pingback: Good Chromatography Practices - SOP & Guideline - Pharma Beginners